Tutorial
How to generate synthetic CRISPR data using DKOsimR?
Abbreviations
KO, knockout; SKO, single knockout; DKO, double knockout; %, percentage; GI, genetic interaction; std. dev., standard deviation.
Introduction
DKOsimR is an R package designed for generating synthetic CRISPR double-knockout screening data. It allows researchers to simulate cell growth dynamics and genetic interactions between gene pairs under controlled library setup and experimental conditions.
This tutorial demonstrates:
Installation
To start running simulation, simply download and install R/RStudio as the first step. You may then install DKOsimR with following commands:
if(!requireNamespace("devtools", quietly = TRUE))
install.packages("devtools")
devtools::install_github("yuegu-phd/DKOsimR", quiet = TRUE)
devtools::install(dependencies = TRUE)
Make sure all required dependencies are installed using devtools::install(dependencies = TRUE).
Then you may simply load the package:
library(DKOsimR)
Graphical Overview of Study Design
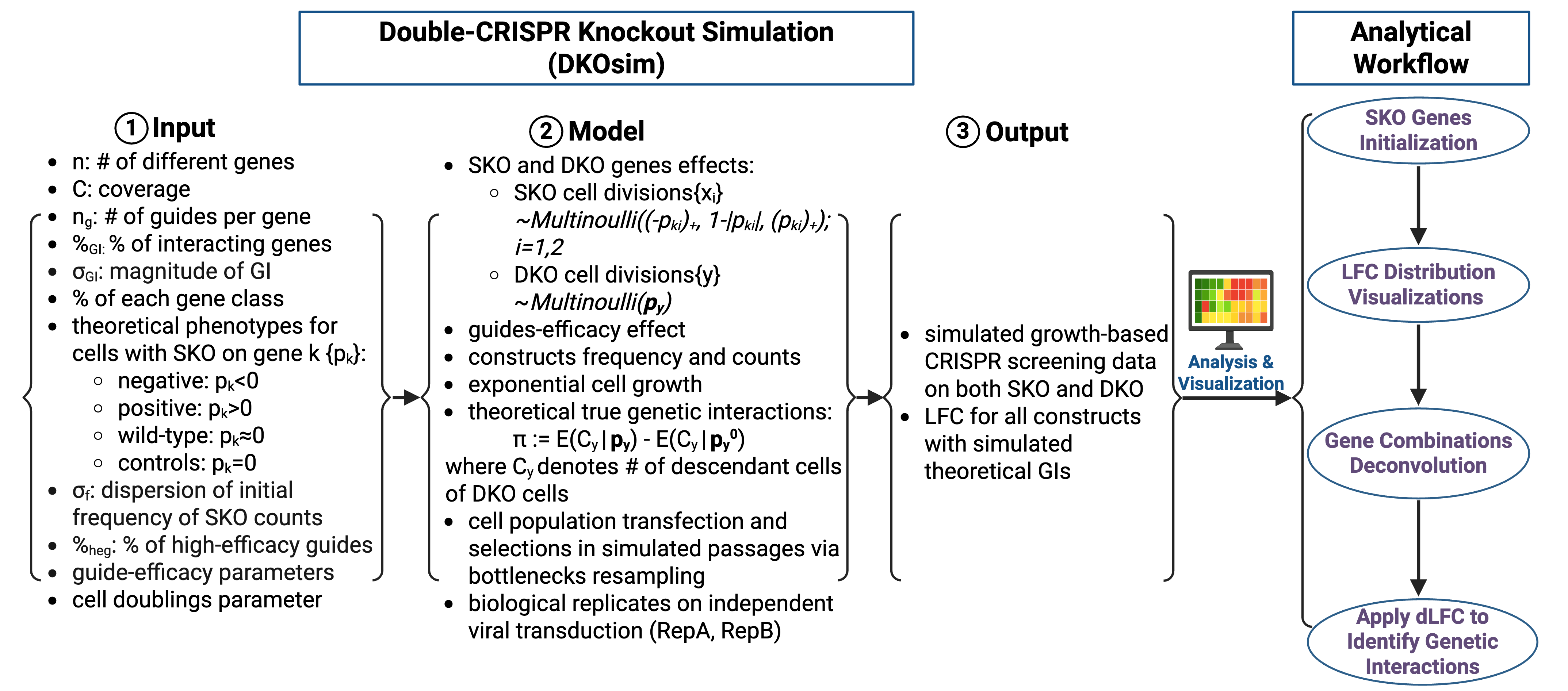
List of Tunable Parameters
Initialized Library Parameters
sample_name: name of the simulation run
coverage: cell representation per guide
n: number of unique single gene
n_guide_g: number of guide per gene
moi: multiplicity of infection - % of cells that are transfected by any virus
sd_freq0: dispersion of initial counts distribution
GI Parameters
p_gi : proportion of interacting gene pairs
sd_gi : std. dev. of re-sampled phenotype with GI presence
Gene Class Parameters
% of theoretical phenotype to each gene class
pt_neg: % negative
pt_pos: % positive
pt_wt: % wild-type
pt_ctrl: % non-targeting control
Mean and std. dev. of theoretical phenotype
mu_neg: mean of negative genes
sd_neg: std. dev. of negative genes
mu_pos: mean of positive genes
sd_pos: std. dev. of positive genes
sd_wt: std. dev. of wild-type genes
Guide Parameters
High-efficacy guides proportion and CRISPR mode
p_high : proportion of high-efficacy guides
mode: CRISPR mode:
use CRISPRn-100%Eff if need 100% effcient guides without randomization
use CRISPRn if need high-efficient guides drawn from distribution
Mean and std. dev. of guide-efficacy
mu_high: mean of high-efficacy guides
sd_high: std. dev of high-efficacy guides
mu_low: mean of low-efficacy guides
sd_low: std. dev of low-efficacy guides
Cell Doublings Parameters
size.bottleneck: bottleneck size - threshold indicating the ceiling of cell growth
n.bottlenecks: number of bottleneck encounters - how many times do we encountering bottlenecks?
n.iterations: number of maximum doubling cycles, by default, we assume a maximum of 30 doublings if we didn’t encounter bottleneck
Randomization Parameter
rseed: values used for random number generator - use same number to control same sets of genes having GI
Miscellaneous
path: path to directory to save outputs of data and logs from simulation
cores_free: number of cores that are left to be free in parallel computing
Quick Start
After loading DKOsimR, to run a simulation with default parameters, you may simply use
dkosim(sample_name = "test", n = 40)
Adjust sample_name and n to name run and initialize number of perturbed genes. Output data will be generated in current working directory.
Alternatively, you may run a simulation in lab approximating mode, by default
dkosim_lab(sample_name = "test_lab", n = 20)
This function applies parameter settings that approximate realistic laboratory data distributions.
Customized Simulation
All tunable parameters may be adjusted by desires in both mode. For example,
dkosim(sample_name="test",
coverage=10,
n=60,
n_guide_g=2,
sd_freq0 = 1/3.29,
moi = 0.3,
p_gi=0.03,
sd_gi=1.5,
p_high=1,
mode="CRISPRn-100%Eff",
pt_neg=0.15,
pt_pos=0.05,
pt_wt=0.75,
pt_ctrl=0.05,
mu_neg=-0.75,
sd_neg=0.1,
mu_pos=0.75,
sd_pos=0.1,
sd_wt=0.25,
size.bottleneck = 2,
n.bottlenecks= 1,
n.iterations = 30,
rseed = 111,
path = ".")
Output data will be generated in current working directory.
Simulation Approximating Laboratory Data
DKOsimR also provides a wrapper function for lab approximating mode to simulate data that resembles real laboratory CRISPR screening datasets:
dkosim_lab(sample_name = "test_lab", n = 20)
This function applies parameter settings that approximate realistic laboratory data distributions.
All parameters can be further customized by users to fit specific experimental setup as desired in both mode, for example:
dkosim(sample_name="test",
coverage=10,
n=60,
n_guide_g=2,
sd_freq0 = 1/3.29,
moi = 0.3,
p_gi=0.03,
sd_gi=1.5,
p_high=1,
mode="CRISPRn-100%Eff",
pt_neg=0.15,
pt_pos=0.05,
pt_wt=0.75,
pt_ctrl=0.05,
mu_neg=-0.75,
sd_neg=0.1,
mu_pos=0.75,
sd_pos=0.1,
sd_wt=0.25,
size.bottleneck = 2,
n.bottlenecks= 1,
n.iterations = 30,
rseed = 111,
path = ".")
Summary
DKOsimR enables researchers to:
generate reproducible synthetic CRISPR screening datasets
benchmark genetic interaction detection methods
evaluate and optimize experimental design parameters
For further information, please refer to the API documentation and the vignettes file (PDF) of DKOsimR R package.