Applying GI Detection Methods on Simulated Data
Delta Log Fold Change (dLFC) Application
Here, we show how we may apply dLFC [1] to calculate GI on simulated data, as an example to analyze the simulation output.
knitr::opts_knit$set(root.dir = getwd())
# read example simulated data
sample_name = "example_data"
pseudo_counts = 5e-07
repA_path <- here::here("data", paste0(sample_name, "_repA.csv"))
repB_path <- here::here("data", paste0(sample_name, "_repB.csv"))
# each simulation is containing two replicates A and B, and we take average of the LFC for analysis
cell_lib_guide2_A = read.csv(repA_path) %>%
dplyr::select(-X)
cell_lib_guide2_B = read.csv(repB_path) %>%
dplyr::select(-X) %>%
dplyr::select(guide_id, counts_guide_t0,
counts_guide_t1, counts_guide_t2,
rel_freq_guide_t0, rel_freq_guide_t2, LFC)
cell_lib_guide2 = merge(cell_lib_guide2_A, cell_lib_guide2_B, by = "guide_id") %>%
dplyr::rename(counts_guide_t0_1 = counts_guide_t0.x,
counts_guide_t0_2 = counts_guide_t0.y,
counts_guide_t1_1 = counts_guide_t1.x,
counts_guide_t1_2 = counts_guide_t1.y,
counts_guide_t2_1 = counts_guide_t2.x,
counts_guide_t2_2 = counts_guide_t2.y,
rel_freq_guide_t0_1 = rel_freq_guide_t0.x,
rel_freq_guide_t0_2 = rel_freq_guide_t0.y,
rel_freq_guide_t2_1 = rel_freq_guide_t2.x,
rel_freq_guide_t2_2 = rel_freq_guide_t2.y,
LFC_t2_1 = LFC.x,
LFC_t2_2 = LFC.y) %>%
mutate(guide1_type = ifelse(guide1_type == 1, "high", "low"),
guide2_type = ifelse(guide2_type == 1, "high", "low"),
construct_type = ifelse(is.na(gene2_behavior), gene1_behavior,
paste0(gene1_behavior, " + ", gene2_behavior)),
LFC_t2_1 = log2(((rel_freq_guide_t2_1 + pseudo_counts) /
(rel_freq_guide_t0_1 + pseudo_counts))),
LFC_t2_2 = log2(((rel_freq_guide_t2_2 + pseudo_counts) /
(rel_freq_guide_t0_2 + pseudo_counts))))%>%
mutate(LFC = (LFC_t2_1 + LFC_t2_2)/2) # aggregate the LFC between 2 replicates by averaging
# read the simulated data and prepare sample name
sim_data = cell_lib_guide2 %>%
mutate(construct_type = ifelse(KO_type == "SKO", "SKO",
ifelse(gene1_behavior == "Non-Targeting Control"
| gene2_behavior == "Non-Targeting Control", "SKO", "DKO" )))
nc_gene = unique(as.character(filter(sim_data, gene1_behavior == "Non-Targeting Control")$gene1))
# calculate single mutant/knockout fitness
gene_SMF = sim_data %>%
filter(construct_type == "SKO", gene1_behavior != "Non-Targeting Control") %>%
group_by(gene1) %>%
dplyr::summarise(SMF = mean(LFC))
# calculate mean LFC for gene pairs that don't contain control
gene_LFC = sim_data %>%
filter(construct_type == "DKO") %>%
group_by(gene_pair_id) %>%
dplyr::summarise(LFC_mean = mean(LFC))
# calculate dLFC = LFC_mean - (SMF1 + SMF2)
sim_data_dLFC = left_join(sim_data, gene_SMF, by = "gene1") %>%
dplyr::rename(gene1_SMF = SMF) %>%
left_join(gene_SMF, by = c("gene2"="gene1")) %>%
dplyr::rename(gene2_SMF = SMF) %>%
left_join(gene_LFC, by = "gene_pair_id") %>%
mutate(gene1_SMF = ifelse(is.na(gene1_SMF), 0, gene1_SMF),
gene2_SMF = ifelse(is.na(gene2_SMF), 0, gene2_SMF),
LFC_mean = ifelse(construct_type == "SKO",
ifelse(gene1_SMF == 0, gene2_SMF, gene1_SMF), LFC_mean)) %>%
mutate(dLFC = LFC_mean - (gene1_SMF+gene2_SMF))
# add data frame for the non-control group
control = sim_data_dLFC %>% filter(gene1 %in% nc_gene | gene2 %in% nc_gene)
sim_dLFC_noncontrol = sim_data_dLFC %>% anti_join(control, by = "gene1_gene2_id")
# add data frame for the non-interacting group
control0 = sim_data_dLFC %>% filter(interaction_gene == 0)
sim_dLFC_noncontrol0 = sim_data_dLFC %>% anti_join(control0, by = "gene1_gene2_id")
# compute correlation and p-values
cor_test_all <- cor.test(sim_data_dLFC$interaction_gene,
sim_data_dLFC$dLFC, method = "pearson")
cor_test_noncontrol <- cor.test(sim_dLFC_noncontrol$interaction_gene,
sim_dLFC_noncontrol$dLFC, method = "pearson")
cor_test_noncontrol0 <- cor.test(sim_dLFC_noncontrol0$interaction_gene,
sim_dLFC_noncontrol0$dLFC, method = "pearson")
# show correlation value
print(cor_test_all)
print(cor_test_noncontrol)
print(cor_test_noncontrol0)
Visualizations
Then you may visualize the calculated GI from dLFC on scatterplots. An example code for plotting might be constructed as follows:
library(ggplot2)
library(ggtext)
# define a function to format p-values if p-values are extremely small
format_p <- function(pval) {
if (is.na(pval)) return("NA")
if (pval < 1e-16) {
return("< 1e-16")
} else {
return(formatC(pval, format = "e", digits = 2))
}
}
# plot the scatterplot
xlim = c(-8,8)
ylim = c(-20,20)
theme_text = theme(
plot.title = element_markdown(size = 8, face = "bold"), # Title size
axis.title.x = element_text(size = 10), # X-axis title
axis.title.y = element_text(size = 10), # Y-axis title
axis.text.x = element_text(size = 10), # X-axis text
axis.text.y = element_text(size = 10), # Y-axis text
legend.title = element_text(size = 10), # Legend title
legend.text = element_text(size = 10), # Legend text
strip.text = element_text(size = 15) # Facet labels
)
scatterplot_dlfc = ggplot(sim_data_dLFC, aes(x = interaction_gene, y = dLFC)) +
geom_point(alpha = 0.7, color = "black") +
geom_point(data = control0, aes(x = interaction_gene, y = dLFC),
size = 3, shape = 17, color = "blue", fill = "yellow", stroke = 1.5) +
geom_point(data = control, aes(x = interaction_gene, y = dLFC),
size = 3, shape = 17, color = "#f76d84", fill = "yellow", stroke = 1.5) +
# Regression line for all data
geom_smooth(method = "lm", col = "black", se = FALSE, linetype = "solid") +
# Regression line for non-control data
geom_smooth(data = sim_dLFC_noncontrol, aes(x = interaction_gene, y = dLFC),
method = "lm", col = "#f76d84", se = FALSE, linetype = "dashed") +
# Regression line for non-zero genetic interactions data
geom_smooth(data = sim_dLFC_noncontrol0, aes(x = interaction_gene, y = dLFC),
method = "lm", col = "blue", se = FALSE, linetype = "dashed") +
ggtitle(paste0(
"Scatter Plot for dLFC vs. Simulated GI:<br>",
"Correlation r = ", round(cor_test_all$estimate, 3),
", *p* ", format_p(cor_test_all$p.value), "<br>",
"Correlation r (w/o non-targeting controls) = <span style='color:red;'>", round(cor_test_noncontrol$estimate, 3),
"</span>, *p* ", format_p(cor_test_noncontrol$p.value), "<br>",
"Correlation r (w/o non-interacting gene pairs) = <span style='color:#0404a0;'>", round(cor_test_noncontrol0$estimate, 3),
"</span>, *p* ", format_p(cor_test_noncontrol0$p.value))) +
labs(color = "Gene-gene Interaction Type") +
guides(color = guide_legend(override.aes = list(size = 5))) +
ylim(ylim[1],ylim[2])+
xlim(xlim[1],xlim[2])+
theme_minimal() +
xlab("Simulated Genetic Interaction") +
ylab("Delta Log Fold Change (dLFC)") +
# Add annotation label for control points
annotate("text", x = xlim[1],
y = ylim[2]-3,
label = "PINK: Data w/ gene pairs containing Non-Targeting Control",
color = "#f85470", size = 3, hjust = 0, fontface = "bold") +
# Add annotation label for non-interacting points
annotate("text", x = xlim[1],
y = ylim[2]-1.5,
label = "BLUE: Data w/ non-interacting gene pairs",
color = "blue", size = 3, hjust = 0, fontface = "bold") +
theme_text
# show the plot
options(repr.plot.width = 8, repr.plot.height = 10)
scatterplot_dlfc
The example scatterplot is shown below:
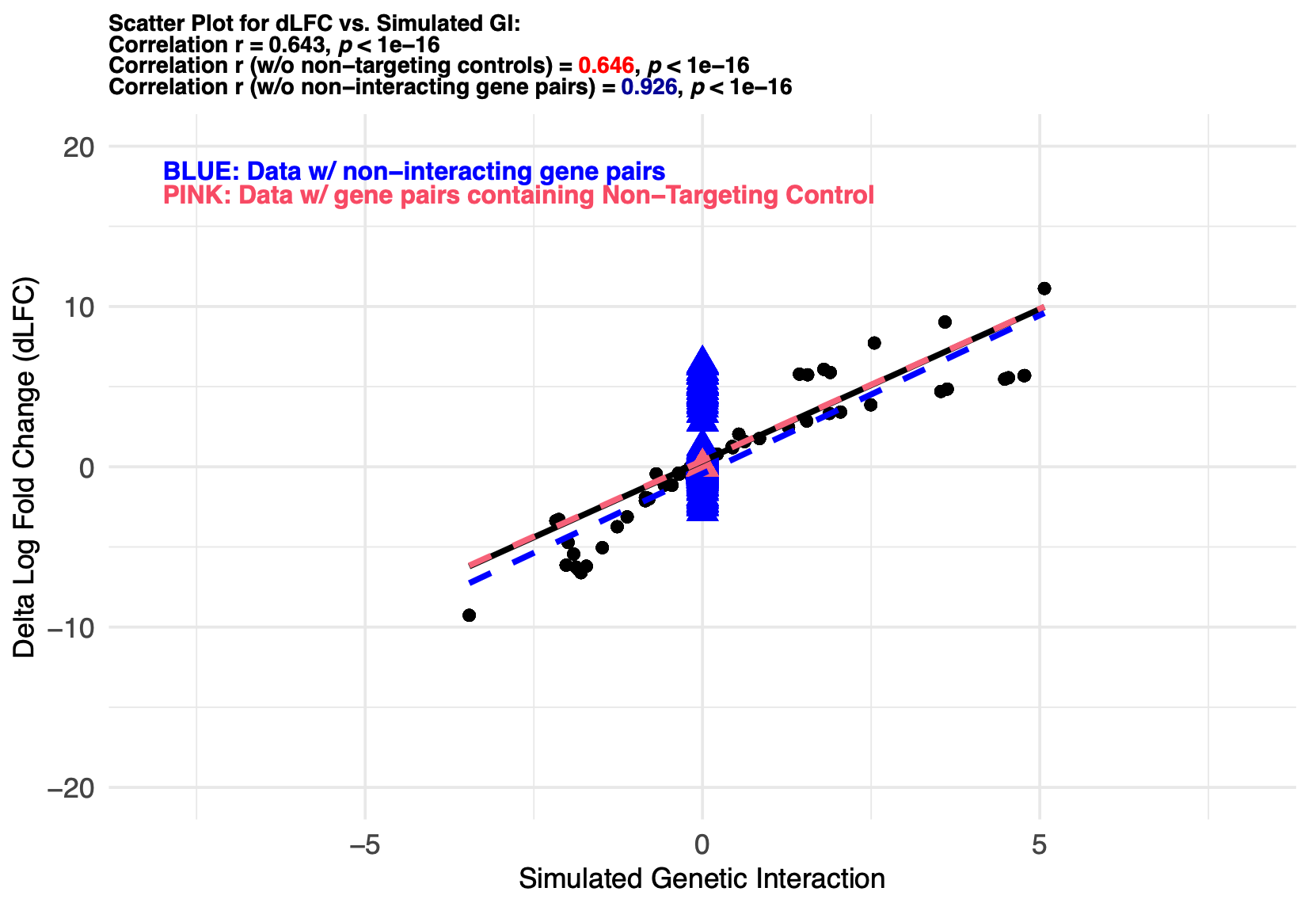
Check relevant details in the pre-built DKOsimR vignettes (PDF) Section 6.
References
[1] Dede, M., McLaughlin, M., Kim, E. et al. Multiplex enCas12a screens detect functional buffering among paralogs otherwise masked in monogenic Cas9 knockout screens. Genome Biol 21, 262 (2020). https://doi.org/10.1186/s13059-020-02173-2.